-
제약산업에서 AI의 활용-1최신 바이오 기술에 관한 지견 2022. 1. 19. 16:15
코로나 바이러스 치료제 개발이 늦어지면서 신약 개발에서 AI의 중요성이 점점 커지고 있다. AI의 활용 가능성을 알아보기 위해 다음 전문을 참고하였다.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7577280/

Artificial intelligence in drug discovery and development, Drug Discov Today.  2021 Jan; 26(1): 80–93. 제약산업에서 AI는 다음과 같은 분야에 활용가능한데, 이 중 몇가지에 대해서 알아보도록 할 것이다.

Artificial intelligence in drug discovery and development, Drug Discov Today.  2021 Jan; 26(1): 80–93. 1) Bioinformatics
신약을 개발하기 위해서 무엇을 가장 먼저 해야 할까? 물론, 이 질문에 대한 명확한 정답은 없다. 사람마다 접근 방식이 다르기 때문이다. 그러나 세포 생물학적인 관점에서 접근을 하자면, 특정 질병을 일으키는 단백질을 찾고, 해당 단백질에 작용하는 약물을 찾아 정상적인 상태로 바꿔 놓으면 질병이 치료된다는 관점에서 신약 개발을 시작할 수 있다.
그렇다면, 이러한 약물의 타겟은 어떻게 찾는 것일까?
인간 게놈프로젝트가 진행되면서 우리는 인체의 모든 단백질 서열에 대해서 알게되었다. 각 단백질들은 서로 영향을 주고 받는데, 이러한 단백질간에 신호전달 과정 연구가 축적되고, 이러한 신호전달 과정들의 상관관계를 하나의 복잡한 회로판에 한데 모아놓았다. 이 후, AI로 하여금 머신러닝을 통해 생물학적인 경로들을 습득하게 했고, 이를 통해 특정 단백질이 사라지거나 억제되었을 때 체내에서 어떤 영향이 일어나는지 예측할 수 있게 되었다. 그러나, 이렇게 AI를 이용해 예측한 결과 값은 Validation이 안된다는 치명적인 문제점을 갖고 있다. 따라서, 결국 실제 세포 실험을 통해, AI에서 예측한 결과가 실제 결과와 동일한지 다시 확인하는 과정을 거쳐야 한다.
또한, Bioinformatics를 이용하여, 약물 재창출(Drug Repurposing)을 해낼 수 있다. 약물 투여 후, 우리 몸에서 유전자 발현 패턴의 변화와 질병 상태에서 우리 몸의 유전자 발현 패턴 변화를 비교하여 약물 재창출을 이루어 낸다.
실제로 코로나 19 바이러스의 치료제 개발 시, 위와 같은 기법을 통해 약물 재창출을 하려는 노력이 이루어졌었다. 아래는 관련 논문 링크이다.
https://www.nature.com/articles/s41598-021-91625-1#Sec2

2) AI - based Drug Discovery
가. Virtual Screening
virtual screening은 CADD(computer aided drug discovery) 방법들을 종합해서 신약 개발에 성공할 가능성이 높은 화합물들을 사전에 필터링 하는 과정을 말한다. 조금 더 자세히 이야기 하자면, 타겟 단백질의 구조가 NMR 혹은 X-ray를 통해 밝혀져 있는 경우 단백질 구조를 가상의 공간에 놓고, 다양한 화합물간에 어떠한 상호작용을 하는지 컴퓨터를 통해 계산해보고(molecular docking 기반) 최적화 약물을 찾는 과정을 말한다. 이러한 과정에서 AI를 활용한다면, LEAD 화합물이 될 수 없는 약물들을 빠르게 제거하는 것이 가능해지므로 많은 시간과 비용을 아낄 수 있다. (대표적인 약물 디자인 알고리즘으로는 coulomb matrices and molecular fingerprint recognition이 있으며, 해당 디자인을 통해 물리화학적, 독성학적 profile을 좋게 만들 수 있다.)
단백질의 구조를 모르지만, 결합하는 ligand 구조만 알고 있는 경우에 QSAR을 통해 약물 구조를 합성하기도 하는데, 이러한 경우에 사용 가능한 AI-based QSAR 모델은 linear discriminant analysis (LDA), support vector machines (SVMs), random forest (RF) and decision trees, 등이 있다.
이렇게 신약 후보 물질을 찾으면 바로 신약이 되는 것일까? 물리화학적 성질, 생체이용률, 독성(안전성) 등 이외에도 다양하게 고려해야 할 부분이 많다. 이러한 부분도 AI를 통해 접근하고 있다.
나. 신약의 물리화학적 성질 예측
용해도, 분배 계수(logP, Octanol과 물에 대한 화합물의 분배계수를 통해 지질친화도를 평가할 수 있기 때문에 신약개발에서 중요하게 보는 물리화학적 성질이다. 이온화되지 않은 화합물의 경우 log P로 표현하고, 이온화된 화합물의 경우 log D로 표기한다.), 이온화 정도(degree of ionization), 등은 약물의 체내 투과력을 결정하기 때문에 신약 개발에 있어서 중요한 역할을 한다. 이러한 물리화학적 지표를 예측하기 위해 Quantitative structure-activity relationship (QSAR)-based computational model이 나타나게 되었는데, 이는 예측의 정확도가 떨어졌다. 그러나 2012년부터 Merck 社는 QSAR 모델에 AI 딥러닝 기술을 접목시켜 신약 후보군의 absorption, distribution, metabolism, excretion, and toxicity (ADMET) data 예측의 정확도를 높였다. (ALGOPS program와 Neuronal network 기반의 ADMET 예측기는 다양한 compound의 용해도와 지용성을 예측하는데 사용되었다.)
다. 신약의 bioactivity 예측
약물이 특정한 효능을 내려면, 수용체와 특정한 상호작용 혹은 친화도를 가져야 한다. 그러므로, Drug-target interaction에 있어서 drug target binding affinity (DTBA)가 중요하다.
AI를 이용한 bioactivity는 target 단백질의 chemical moiety와 약물의 chemical moiety 사이의 특징을 인식하여 약물 상호작용을 확인하는 Feature based와 동일한 타겟에 결합하는 약물과의 유사성을 통해 상호작용을 예측하는 Similarity based affinity가 있다. ex) ChemMapper and the similarity ensemble approach (SEA)KronRLS(ML based), SimBoost(regression trees 이용) 그러나 최근에 ML보다 DL 기반의 신약 bioactivity 예측이 보다 낫다고 밝혀지고 있음. 왜냐하면 DL은 ML과는 다르게 3D 단백질 구조를 알고 있는지의 유무와 상관 없이 network based method를 이용하기 때문이다. DeepDTA, PADME, WideDTA, and DeepAffinity - DL 을 이용해 DTBA 예측
XenoSite, FAME, and SMARTCyp - 약물의 metabolism 예측
CypRules, MetaSite, MetaPred, SMARTCyp, and WhichCyp - CYP450 중 어떤 isoform에 의해 대사되는지 예측 가능
라. 신약의 독성 예측
LimTox, pkCSM, admetSAR, and Toxtree
최초로 AI로 독성을 예측하고자 했던 것은 NIH, EPA, FDA가 설립한 Tox21 Data Challenge가 있다.

AI Tool 신약의 화학 구조 역시 AI를 이용해서 예측할 수 있다.
마. AI를 이용한 약물 구조 디자인
ㄱ) 타겟 단백질 구조 예측
AlphaFold: DNN(deep neural networks) 기반, 단백질의 3차 구조를 예측하게 해주는 AI tool, 근접한 아미노산과 상응하는 펩티드 결합의 angle을 분석하는데 사용함.
AlQurashi: RNN 기반, Alphafold 보다 더 빠르게 단백질 구조를 예측 가능, 그러나 Alphafold가 보다 더 정확하게 구조를 예측함.
MATLAB: 단백질의 2차원 구조를 예측함. 구조 예측 정확도는 62.72%
2차원 구조 예측도가 62.72%이므로, 실제 3차원 구조 예측 정확도는 이보다 낮을 것으로 조심스럽게 예측해볼 수 있다.
ㄴ) 약물-단백질 상호작용
iDrug-GPCR, iDrug-Chl(이온 채널), iDrug-Enz(효소), iDrug-NR(핵 수용체): 약물-단백질 상호작용 파악 가능하다.
또한 이러한 세포 내에서 일어나는 network를 기반으로 하여 Deep learning을 시킨 deepDTnet는, topoisomerase inhibitor인 topotecan이 human retinoic acid receptor-related orphan receptor-gamma t (ROR-γt)를 차단한다는 것을 밝혀냈고, 이를 통해 현재 다발성 경화증을 적응증으로 약물 재창출을 시도하고 있다. 이미 기존에 있던 약을 새로운 적응증으로 시장에 다시 launching 하는 것(US$8.4 million)은 신규물질을 개발하여 시장에 출시하는 것(∼US$41.3 million)보다 비용적인 면에서 많은 이익이 있다.
Off target을 찾을 수 있다. - 약물 재창출 및 off target adverse effect 예측 가능
- Self-organizing maps (SOMs): unsupervised ML로써, 생물학적 활성을 갖는 것으로 알려진 ligand와 타겟 단백질 간의 결합 정도를 훈련시켜, 물질의 새로운 off target을 찾는 ligand based approach를 사용한다. 이 때 해당 off target effect가 치료 효과가 있으면, 약물 재창출, 부작용으로 이어지면, off target adverse effect를 예측하는 것으로 해당 기술을 사용할 수 있다.
- Bayesian classifiers와 SEA algorithms은 약물의 약물학적 profile과 그들이 가능한 타겟 단백질 간의 link를 규명하는데 사용가능하다.
- KinomeX: DNN 기반으로, 화학 구조와 kinase 간의 polypharamcology 관계를 확인하는 AI platform, 14000개의 생물학적 활성 데이터와 300개 이상의 kinase를 기반으로 학습을 시켜, Kinase family/subfamiliy 관련 신규 화학물질 design 시 도움을 준다. 해당 platform으로 NVP-BHG712이라는 물질의 primary target과 off target effect을 밝혀냄.
- Ligand Express: Cyclica의 cloud based proteome screening AI platform으로써, small molecule과 결합 가능한 단백질을 예측 가능하게 해준다. 따라서 이를 통해 off target을 유추할 수 있고, adverse effect을 예측할 수 있다.
ㄷ) AI를 활용한 신규 약물 구조 디자인 (AI in de novo drug design)
- Merck의 Synthia: 8개의 의학적으로 필수적인 타겟 단백질에 결합할 수 있는 물질을 합성하는 적합한 방법을 제공하는 능력을 가진 AI platform이다. 전문 유기 화학자에 의해 10만개 이상의 반응 규칙을 코딩하여 역합성 소프트웨어를 개발하였다.
- Reinforcement Learning for Structural Evolution strategy for de novodrug synthesis: 딥러닝과 Reinforcement Learning(어떤 환경 안에서 정의된 에이전트가 현재의 상태를 인식하여, 선택 가능한 행동들 중 보상을 최대화하는 행동 혹은 행동 순서를 선택하는 방법)을 기반으로 simplified molecular-input line-entry system (SMILES) strings의 화학물질 데이터를 학습시켜 새로운 신약의 화학 구조를 개발하게 함.

AI를 활용하여 약물 구조를 개발하는데 있어서 사실, 고순도의 input data가 방대하게 넣고 Machine Learning을 할수록 신규 화학 물질이 발견될 가능성이 높기 때문에 최근에는 MELLODY라는 컨소시엄을 구성하여 신약 후보물질의 scaffold개발을 같이 하고 있다. MELLODY 프로젝트는 2019년 6월 시작되었으며, 참여 제약 회사는 3년간 해당 연합학습을 통해 나타난 모든 예측 물질들을 자유롭게 사용 가능하다. 각 제약회사가 해당 프로젝트에 제공한 chemical library는 각 제약회사의 data set이기 때문에 private blockchain 기술을 이용해 이를 엔비디아에서 제공한 자체 클러스터에 데이터를 보관합니다. 이후, 이 데이터를 이용해 연합학습을 시킵니다. 이후, 기업의 이익이 가시화 되는 patent한 물질이 나타나는 순간 branch out한다.

https://astrixinc.com/melloddy-consortium-employs-federated-learning-and-blockchain-to-enhance-ai-drug-discovery/ 3) 디지털 가상 세포, 장기, 인체
가. 디지털 세포
가상 공간에서 분자 수준의 상호작용을 미리 살펴봄으로써 화합물의 세포 내 행태를 미리 예측하는 것을 말한다. 이는 NGS 기술의 발달로 인해 유전자의 기능 및 세포 내의 모든 pathway를 알게되고, 이를 수학적으로 modeling하여 하나의 세포를 모사해 만들게 됨으로써 디지털 가상세포라는 개념이 나타나게 되었다. 20년 전 일본 연구진에 의해 유전자가 가장 작은 세포인 M.genitallium을 E-cell로 구현해 낸 것이 현재 디지털 세포의 시초이다.
그러나 아직 해당 기술은 완벽하게 구현되고 있지 않다.
나. 디지털 장기
독일 바이엘 사가 개발한 PBPK model, MOBB라고 하는 소프트웨어가 대표적이다. 몸속에 특정 약물이 들어갔을 때, 어떻게 각 장기별로 분포되고 그것이 특히 간세포에 도달했을 때, 간세포의 어떤 분자와 상호작용을 하고, 어떻게 유전자 전사 조절 과정에 관여하는지, 등을 추적할 수 있도록 만든 소프트웨어이다.
MOBB= 간세포 안에서 어떤 것이 일어나는지 알기 위해 만든 소프트웨어로, 완벽하지 않다. 현재도 새로운 것이 밝혀질 때마다 해당 platform을 업데이트하여 보완해 나가고 있다.
최근 PBPK 모델을 통해 간 흡수 수송체에서 특정 표현형을 가진 환자집단을 식별하거나(Krauss et al. 2013) 환자의 고위험 하위 그룹에서 부작용 발생률을 에측하는데 사용되고 있다. (Lippert 외 2012)
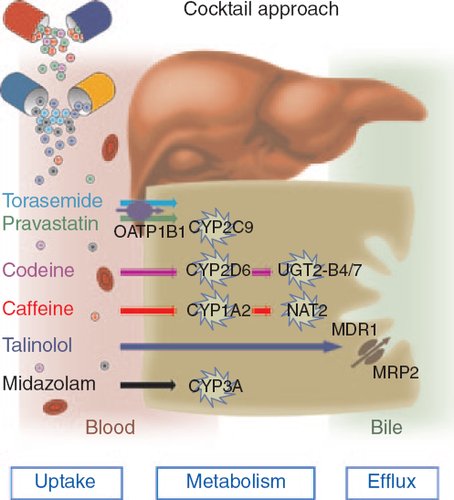
또한 가상 간 네트워크 내에서 6가지 약물 칵테일 요법을 사용하여 마우스와 인간의 간의 해독 활동을 정량화하였다. (Kuepfer et al. 2014 ) 해당 연구들은 모두 2014년, 독일의 Virtual Liver Network에서 연구했다.
https://ascpt.onlinelibrary.wiley.com/doi/full/10.1038/psp.2014.25 다. 디지털 인체
최근 국내에는 CODA라고 불리는 가상 인체를 개발하고 있다.

https://www.biosynergy.re.kr/development-1/view/id/346#u 상황정보가 포함된 생체 내 다 수준 간의 생물학적 관계 정보를 수집 및 통합하여 세포/조직/질병 특이적인 Context-Oriented Directed Association (CODA) repository를 구축하고 이를 통해 네트워크를 만들고, 해당 네트워크에서 특정 약물이 들어갔을 때 어떠한 효능이 나타나는지 예측할 수 있다. 따라서 이를 통해 약물 재창출, 약물 효능 예측, 시너지 복합 약물 후보군 예측 등이 가능해진다.
2편으로 돌아오겠습니다!
'최신 바이오 기술에 관한 지견' 카테고리의 다른 글
Global Big Pharma의 AAV Gene Therapy Biotech M&A/ Gene Therapy 회사 간 비교 (0) 2021.02.07 AAV gene therapies의 최신 동향 및 문제점 (0) 2021.02.06 AAV Gene Therapy 소개 (0) 2021.02.04